
「Avocute」は半経験的量子力学計算 (経験的パラメータを使用した分子の電子状態計算)を行うための軽量で無料の計算化学ソフトウェアです。ダウンロードするだけで、すぐに使いはじめることができます!!

難しい操作や煩雑な操作画面を排除した非常にシンプルなデザインになっているので、計算化学にはじめて取り組む方でも安心して使用できます。Avocuteが少しでも誰かのお役に立てれば幸いです!!
※) 当ブログの連載が書籍になりました!!興味がある方は、お手に取っていただければ幸いです!!
(Amazon購入ページ)(書籍の紹介記事)

【バージョン】 2.0.0
【最終更新日】 2020/11/06
【ダウンロード】
以下のリンクから、最新版のAvocuteをダウンロードすることができます。
【免責】
本ソフトウェアは十分なテストを行っていますが、動作に関してなんらかの保証をするものではなく、いかなる運用結果についても一切の責任を負いません。あらかじめご了承ください。
【アンインストール】
zipファイルを解凍した際に作成されるフォルダごと削除して下さい。本ソフトウェアはユーザーが直接指定しない限りフォルダ外へのファイルの作成はおこないません。また、レジストリの変更も行いません。
【旧版のダウンロード】
過去のバージョンはGitHubより入手できます。
GitHub Avocute
スポンサードリンク
【主な機能 】
・ 計算に必要な入力ファイルの作成と計算の実行
・ 複数の入力ファイルを一括で実行できるバッチ計算用ツール
・ 反応解析に必要な遷移状態を反応物、生成物より計算する自動ツール
・ IRC計算の最終構造、エネルギープロファイルのCSV出力
Avocuteには、分子モデルをグラフィカルに表示する機能はありません。別途、分子モデリングのためのエディターを用意する必要があります。 Avocuteは、分子エディターにAvogadroを使用することを推奨しています。
Avogadroは、以下のページから無料で入手できます。
Avogadro
https://avogadro.cc/
【変更履歴】
・2020/11/06 Ver.2.0.0公開
・MOPAC6をMOPAC6.06へ変更
・mgfファイルの出力に対応(JSmolで分子軌道表示可能)
・メイン画面の[Calculation]にIRCメニュー追加
・メイン画面の[Options]の「VECTORS」を「GRAPH」に変更
・メニューバーのToolsに「IRC Checker」追加
・ARCファイルの読込みに対応
・JSmolのURL修正
・その他、軽微な不具合の修正
・2020/10/15 Ver.1.0.0公開
同梱の関連ファイルを削除したり、ソフトウェア本体をファイル外へ移動したりすると正常に動作しなくなるので注意してください。
Avocute.exeのショートカットを好きな場所に作成して利用することをおすすめします。
非常にシンプルなUIなので説明がなくても問題なく使用できるかと思います。
具体的な使用方法は以下の通りです。説明は、Ver.1.0.0の操作画面を用いて行っています。最新バージョンで追加された項目・機能については、その都度、補足します。
実際に「水分子の構造最適化計算」を例に実行方法を説明します。
構造最適化とは、簡単に言うと「分子の結合角や、結合長を含めた安定な構造を求める計算」のことです。
1)まず、Avogadroを起動し水分子を構築します。水分子の作成方法がわからない場合は以下のページを参照してください。
【関連記事】Avogadroを使ってみよう
http://pc-chem-basics.blog.jp/archives/396177.html
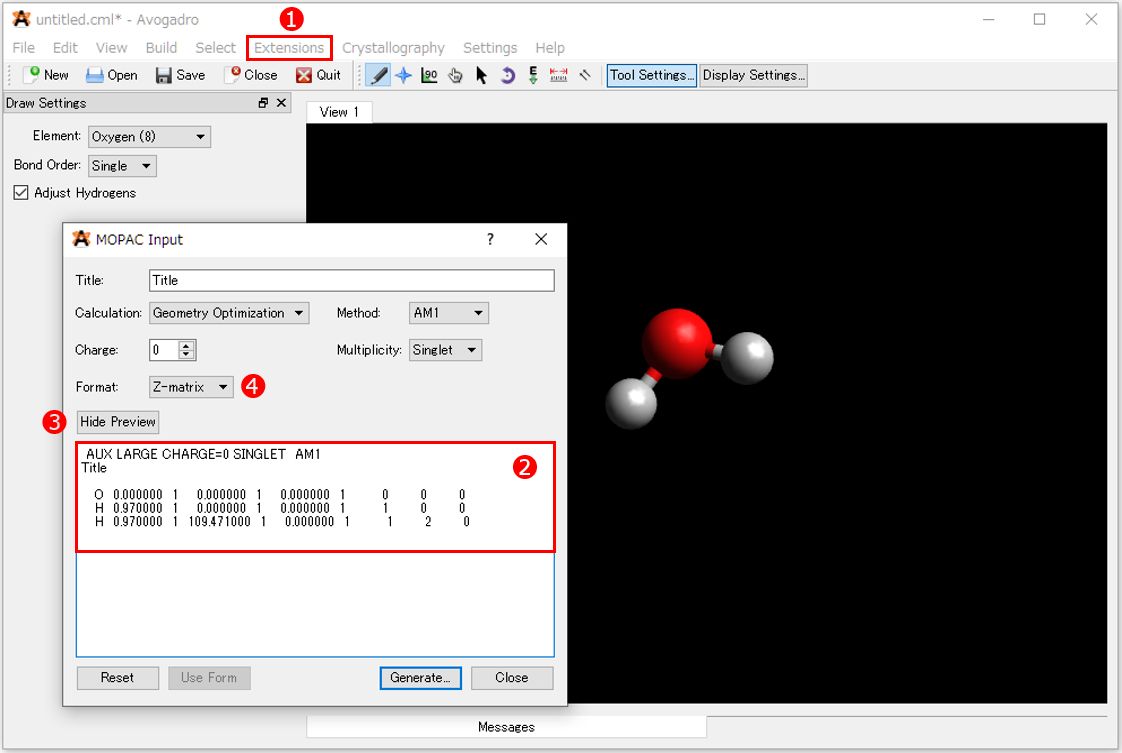
水分子が作成できたらAvogadroのツールバーから①「Extension>MOPAC...」を選択します。表示される「MOPAC Input」画面の②エディット画面のテキストを全て選択してコピーします。
エディット画面のテキストが表示されていない場合は、③[Show Preview]ボタンをクリックすることで表示/非表示(Hide Preview)を切替えることができます。 この画面で計算方法を指定しても当然問題ありません。作成例では、分子座標を④[Z-matrix]に指定し、その他の設定は変更しません。
2)先程コピーしたテキストをAvocuteのメイン画面の②エディット画面(MOPAC Input:)に貼り付けます。 入力ファイルは、最初の行に使用するパラメータモデル(AM1, PM3など)と計算に必要なキーワードを指定します。 2行目と3行目はスペースです(個人的なメモを記入してもOKです)。4行目から、分子座標を入力します。そして、分子座標の最後に空白行という構成になっています。
貼り付けたテキストの最後に一行以上空白行があるか、必ず確認してください(赤枠部分)。これらはMOPACの入力ファイルの形式上の決まり事です。入力ファイルの形式が守られていない場合、計算エラーが出力されるので注意が必要です。
計算手法や分子座標について、さらに詳しく知りたい方は以下のページを参照してください。
【関連記事】
MOPACの入手とインストール方法
http://pc-chem-basics.blog.jp/archives/1496795.html
MOPACを実行してみよう
http://pc-chem-basics.blog.jp/archives/1509833.html
メイン画面のエディターは、直接書き込んだりコピペしたりする他にも、メニューバーの「File>Open Input File...(*.dat)」から既存のファイルを読み込むことが可能です。必要に応じて上手に使い分けてください。
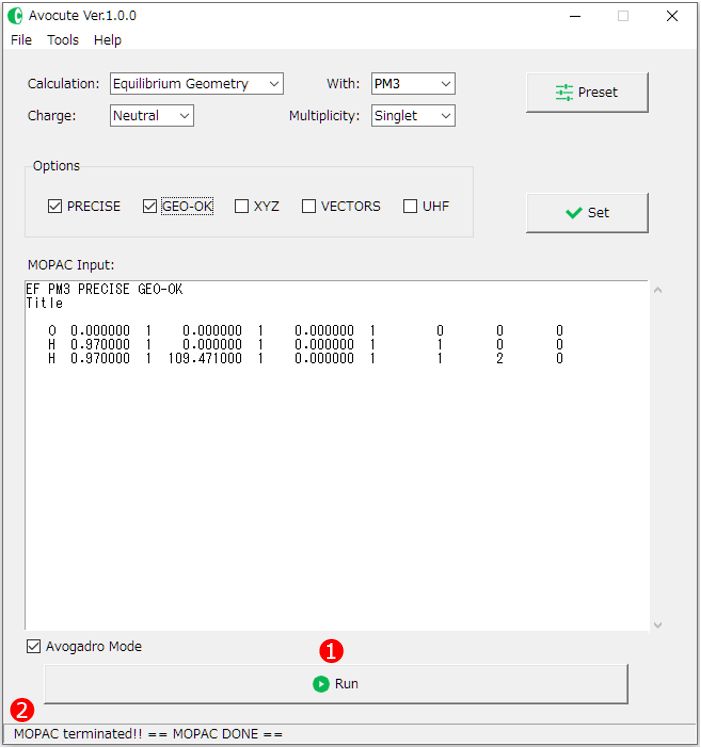
3)このまま実行ボタン[Run]をクリックして計算を実行することもできますが、ここでは設定ボタンで計算方法を変更してみましょう。使用例ではパラメータにPM3を用いて構造最適化計算を行います。つまり、各プルダウンメニュー①は図のように設定します。
CalculationはEquilibrium Geometry (構造最適化)、パラメータ(with)はPM3、電荷(Charge)、スピン多重度(Multiplicity)は中性分子なのでNutral、Singlet(一重項)をそれぞれ選びます。
これら4つは必ず適切に指定する必要があります。
オプション②は必ずしも必要ではありませんが、ここではPRECIESとGEO-OKにチェクを入れてみましょう。オプションに用意されているキーワードの意味は以下の通りです。
PRECISE:収束判定を100倍厳しくする (より厳密な構造最適化を希望する場合)
GEO-OK:原子が異常に接近した場合のチェックを無視する
XYZ: カルテシアン座標系で計算 (内部座標系でエラーが出る場合などに追加)
VECTOR:軌道係数、軌道エネルギーなどを出力ファイルに印字 (Ver.1.0.0のみ。Ver.2.0.0以降もキーワードは使用可能)
UHF:非制限ハートリー・フォック法 (開殻電子系に使用)
GRAPH:JSmolで分子軌道を表示するための書式付ファイル(*.mgf)を生成する (Ver.2.0.0より追加)
最後に③[Set]ボタンをクリックすれば、エディット画面の一行目に設定したキーワードが反映されます。
ちなみに、④[Preset]ボタンをクリックするとお気に入りのキーワードを登録して呼び出すことができます。⑤の入力欄に登録したいキーワードを入力して、保存したい⑥「Save Slot」を選んだ状態で右端の⑤[Save]ボタンをクリックすると登録完了です。⑦[Set]ボタンをクリックすればメイン画面にキーワードを反映することができます。
MOPACにはさらに多くのキーワードが存在します。MOPAC6に対応する全てのキーワードを確認するには、Avocuteのメニューバーの「Help>MOPAC6 Keywords」から参照してください。
メイン画面下、⑧[Avogadro Mode]のチェックが入っている場合は、MOPAC6に対応していないキーワード(AUX:他のプログラムで利用可能な追加情報ファイル*.auxの出力)を自動的に削除して計算します。これは、AvogadroのMOPAC入力ファイルのエディット画面で自動的にAUXが追加されていても、Avocuteでそのまま計算を実行できるようにするためのものです。キーワードAUXは最近のMOPACに実装されたものでMOPAC6には対応していません。
4)最後に、①[Run]ボタンをクリックして計算を実行してみましょう。はじめに入力ファル(*.dat)をどこに保存するかを選択すると、直ちに計算が実行されます。
保存先のディレクトリ名はスペースや特殊な記号などを含むとエラーが出るので、それらを含まない場所やファイル名で保存してください。Avocuteの実行ディレクトリも同様です。
計算が開始すると②ステータスバーの表示が「wait...」になります。計算が終了すると、「MOPAC terminated!!」と表示されます。 計算が正常に終了した場合は、「== MOPAC DONE ==」、エラーが発生している場合は「Error: Check the output!!」と続いて表示されます。
5)計算が正常に終了したら、入力ファイル(*.dat)を保存した場所に同じファイル名で「*.arc」、「*.out」が作成されているはずです。「*.out」が計算結果を記した出力ファイル、「*.arc」が「*.out」の要約を記したファイルになります。いずれも、テキストエディッタで開いて内容を確認することができます。計算エラーの場合は、「*.out」をテキストエディッタで開き出力ファイルに示されているエラーの原因を確認しましょう。
生成する「*.arc」、「*.out」は、すでに同じ名前のファイルが存在する場合、自動で上書きされます。他のツール(Batch Launcher、Auto TS Searcher)でもこの仕様は同様です。
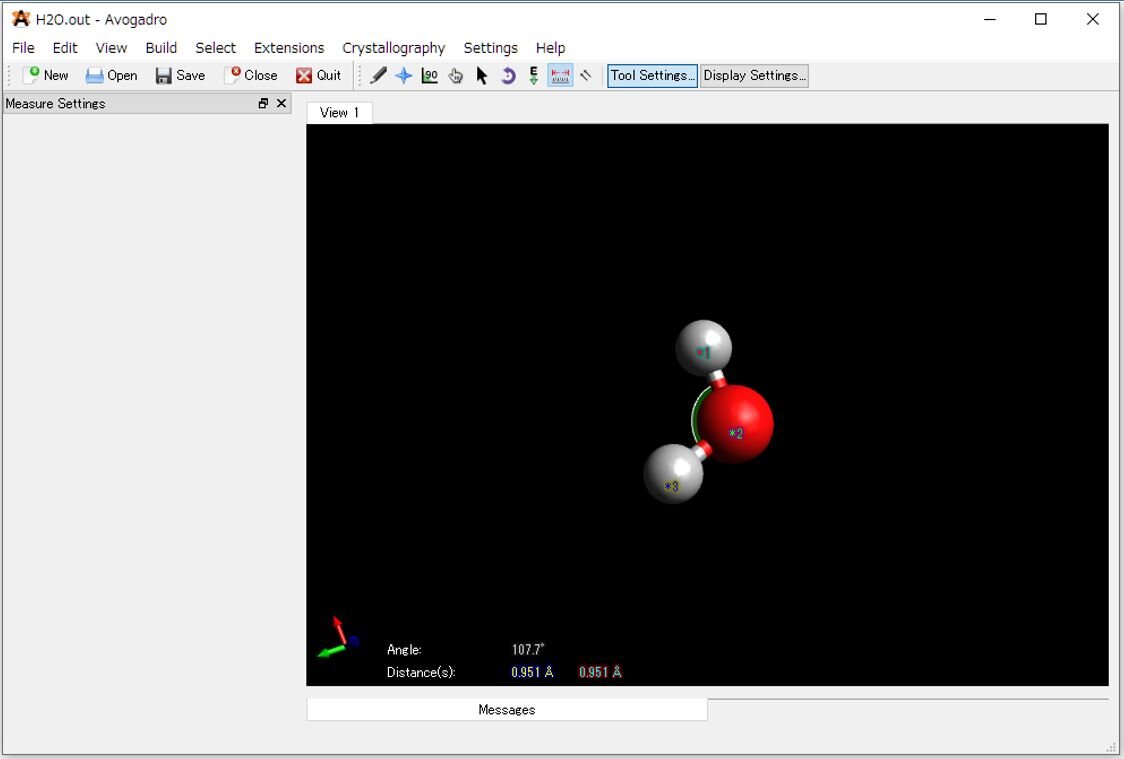
6)出力ファイル(*.out)は、Avogadroのメイン画面にドラックアンドドロップすることで構造最適化した構造を表示することができます。Avogadroを使えば、計算で得られた分子の結合長や結合角などをグラフィカルに確認することができます。
まず、メイン画面のエディターで入力ファイルを作成します。Batch Launcher画面の①[Add]ボタンをクリックすると入力ファル(*.dat)をどこに保存するか選択し、登録完了です。
あとは、同様に計算したいファイルを登録していき②[Run]ボタンをクリックすると計算が順次実行されます。計算が終了すると、Statusが「Pending」から「Done」に変わります。 登録したデータをクリアしたい場合は、③[Reset]ボタンをクリックしてください。
ちなみに、メニューバーの「File>Clear Temporary Files」を実行すると計算時に作成される不要な一時ファイルを削除することができます。通常の計算では自動的に不要な一時ファイルは削除されるので、このメニューを実行する必要はありません。 計算が途中でクラッシュした場合や、計算結果が以前の計算結果に影響を与えている可能性がある場合、またはAvocuteフォルダ内に不要なファイルが蓄積した場合などに実行してください。
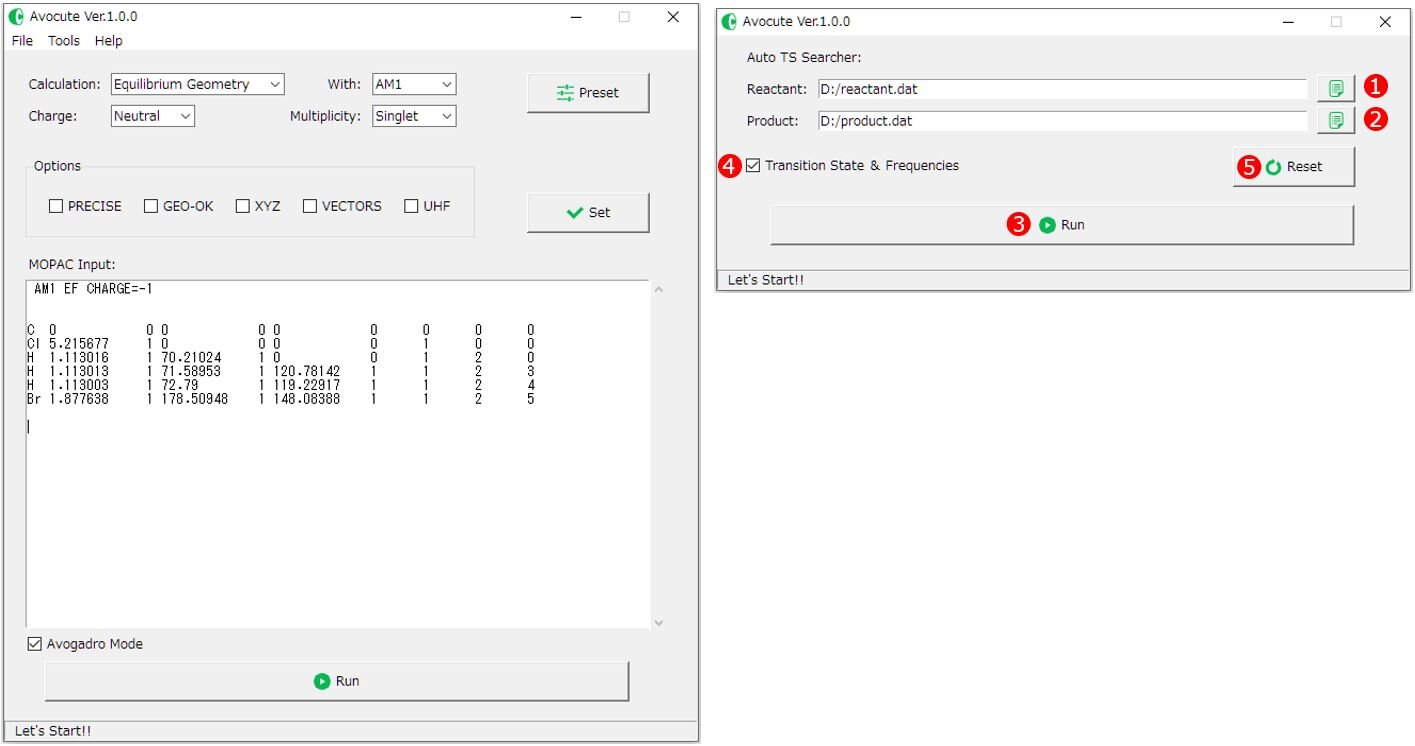
1)まず、メイン画面で反応物の構造最適化の入力ファイルを作成します。Auto TS Searcher画面の①Reactant(反応物) 登録ボタンをクリックし入力ファル(*.dat)をどこに保存するか選択し、登録完了です。同様にメイン画面のエディターで生成物の入力ファイルを作成したら②Product(生成物)登録ボタンをクリックし登録します。
2)この使用例では、Avocuteに同梱されている「SampleFile」フォルダの中から「product.dat」と「reactant.dat」を用いて計算を実行します。
サンプルファイルは塩化メチルと臭化物イオンの求核置換反応における反応物と生成物です。 登録が完了したら③[Run]ボタンをクリックして計算を実行しましょう。
④[Transition Stae & Frequencies]にチェックがある状態(デフォルト)では、SADDL計算後に遷移状態の最適化、および振動解析を実行します。チェックを外すとSADDLE計算で終了します。必要に応じて使い分けてください。
なお、Reactant(反応物)とProduct(生成物)の構造最適化後に行われる、SADDLE計算、遷移状態の最適化(TS)、振動解析(FORCE)で用いられるオプションのキーワードは、Reactant(反応物)の入力ファイル中に含まれるキーワードをそのまま継承します。
もし、登録したファイルを解除したい場合は、⑤[Reset]ボタンをクリックしてください。
3)計算が開始するとステータスバーの表示が「wait...」になります。計算が終了すると、「MOPAC terminated!!」と表示されます。 計算が正常に終了した場合は、「== MOPAC DONE ==」と続いて表示されます。
4)計算が正常に終了したら、反応物の入力ファイル(*.dat)を保存した場所に同じファイル名で「*.arc」、「*.out」が作成されているはずです(振動解析(キーワード:FORCE)の計算ではarcファイルは作成されません)。
遷移状態の候補化合物(SADDLE)は「反応物のファイル名_saddle」、その最適化計算(TS)は「反応物のファイル名_ts」、その振動解析は「反応物のファイル名_frq」というファイル名で保存されます。
5)出力ファイル(reactant_ts.out)を、Avogadroの画面にドラックアンドドロップすると計算で求めた構造をグラフィカルに表示することができます。
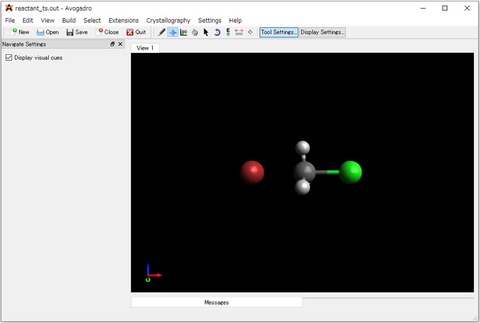
6)振動解析の結果(*_frq.out)はAvogadroで表示できません。グラフィカルに結果を確認したい場合は、Avocuteのメニューバー「Tools」から「JSmol」を選択するとWebブラウザでJSmolにアクセスできます。
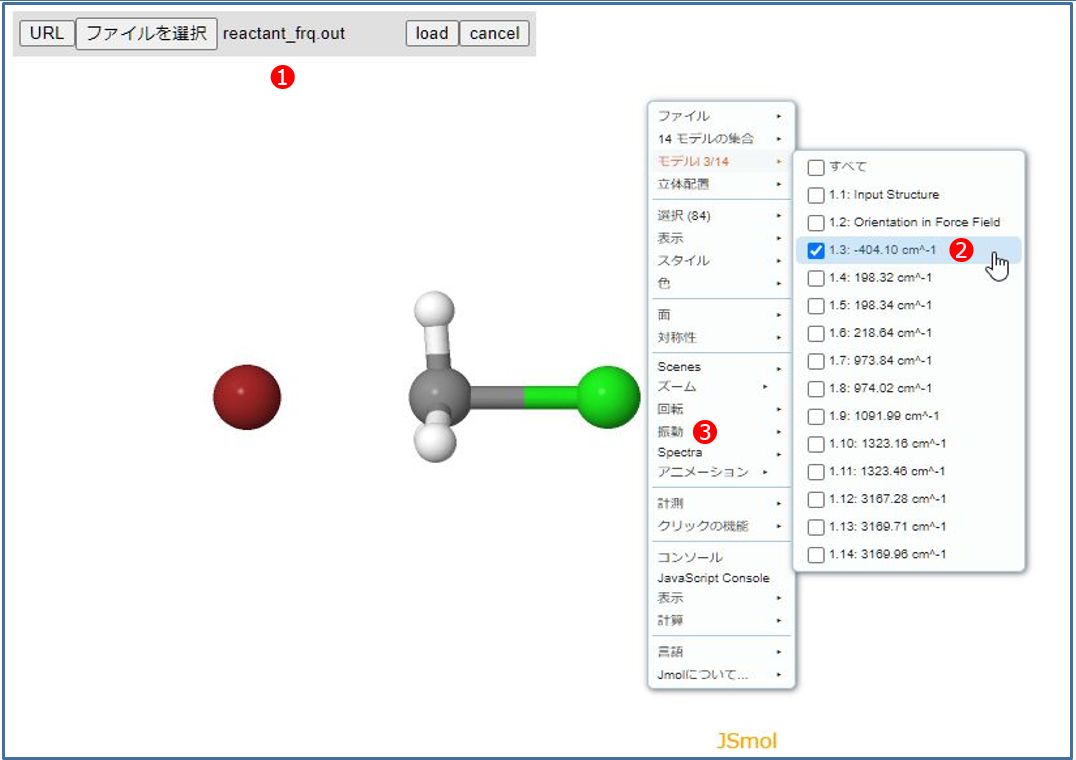
ファイル選択メニュー①が表示されるので「reactant_frq.out」を選択して[load]ボタンをクリックすると目的の分子構造が表示されるはずです。
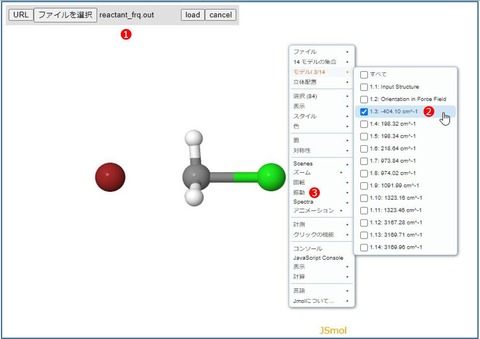
次に右クリックメニューの②「モデル」から目的の振動を選択します。ここでは、遷移状態の確認なので唯一存在する虚数振動(マイナスの振動数)を選択します。 最後に③「振動」をオンにすれば振動モードに対応したアニメーションが表示されます。
最後に、SADDLE計算をする際に注意しておきたいのは、内部座標系を用いる場合は反応物と生成物の各結合情報が一致していなければならないことです。例えば、反応前後で各原子に割り当てられる番号が異なっていたり、内部座標で定義されている参照原子との関係が異なったりしていると、エラーが出力されるので十分注意しておきましょう。

上図は、Avocuteに同梱されている「SampleFile」フォルダの中から「phenol.mgf」のHOMOを表示させた様子です。JSmolの右クリックメニューの「面>分子軌道」より目的の軌道を選択するだけです。
IRC計算とは、得られた遷移状態が反応物と生成物を結ぶかどうかを確認するために用いられます。先述の「Auto TS Searcher」で得られた、遷移状態を元にIRC計算を行いながら一連の操作方法を説明します。
1) ここでは、Avocuteに同梱されている「SampleFile」フォルダの中から、「reactant_ts.arc」を用いて説明します。「reactant_ts.arc」には、計算で得られた、塩化メチルと臭化物イオンの求核置換反応における遷移状態の分子座標が含まれています。
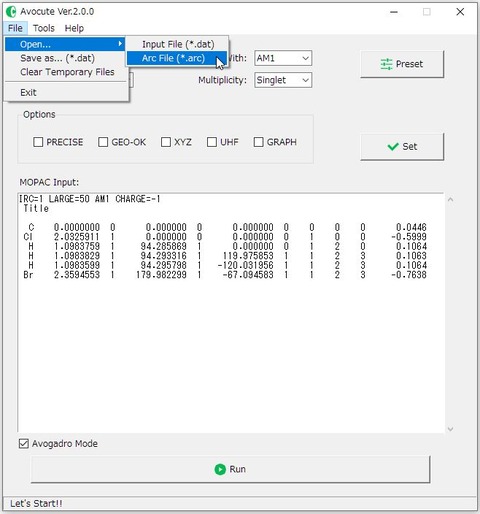
メニューバーの「Fire >Open... >Arc Fire(*.arc)」からファイルを読み込みましょう。キーワードは、上図のように「IRC=1 LARGE=50 AM1 CHARGE=-1」と設定します。メイン画面の[Calculation:]のプルダウンメニューから「IRC」を選択した場合、「IRC=1 LARGE=50」がデフォルトです。
キーワード「IRC=1」は、虚の振動数をもつ基準振動の方向に構造変化させることを、「IRC=-1」は逆方向へ変化させることを意味します。遷移状態から、反応物と生成物への両方の確認が必要なので、「IRC=1」の計算が終了したら、「IRC=-1」に変更して計算を実行しましょう。ちなみに、「LARGE=50」は、50回ごとにIRC上の構造を出力する事を指定しています。「CHARGE=-1」は、モデル全体の電荷を指定しています。
2) 入力ファイルの作成が終了したら、メイン画面の[Run]ボタンをクリックして計算を行いましょう。計算が終了すると、保存したファイル名の「*.out」が出力されるはずです。
3) メニューバーの「Tools >IRC Checker」をクリックし、先程の計算で出力された「*.out」ファイルを読み込みましょう。IRC計算以外のOutputファイルや、計算が正常に終了していないファイルを読み込むとエラーが出力されます。
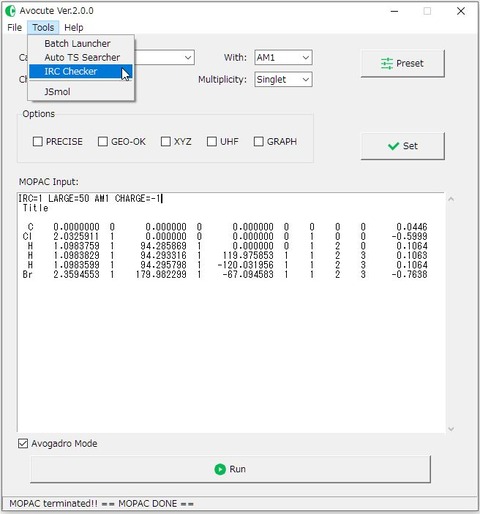
すべての処理が正常に終了すると、下図のように「Successfully terminated!!」というメッセージボックスが表示されます。また、読み込んだ「*.out」ファイルと同じ場所に、「*_ircf.out」「*_ircf_plot.csv」の2つのファイルが出力されます。
難しい操作や煩雑な操作画面を排除した非常にシンプルなデザインになっているので、計算化学にはじめて取り組む方でも安心して使用できます。Avocuteが少しでも誰かのお役に立てれば幸いです!!
※) 当ブログの連載が書籍になりました!!興味がある方は、お手に取っていただければ幸いです!!
(Amazon購入ページ)(書籍の紹介記事)
Avocuteのダウンロード
【動作環境】 Windows 7 / 8 / 10【バージョン】 2.0.0
【最終更新日】 2020/11/06
【ダウンロード】
以下のリンクから、最新版のAvocuteをダウンロードすることができます。

【免責】
本ソフトウェアは十分なテストを行っていますが、動作に関してなんらかの保証をするものではなく、いかなる運用結果についても一切の責任を負いません。あらかじめご了承ください。
【アンインストール】
zipファイルを解凍した際に作成されるフォルダごと削除して下さい。本ソフトウェアはユーザーが直接指定しない限りフォルダ外へのファイルの作成はおこないません。また、レジストリの変更も行いません。
【旧版のダウンロード】
過去のバージョンはGitHubより入手できます。
GitHub Avocute
スポンサードリンク
Avocuteとは?
Avocute本体は、同梱のMOPAC6(半経験的量子力学計算のソフトウェア)を操作するためのUI(ユーザーインターフェイス)として機能します。主な機能は以下の通りです。【主な機能 】
・ 計算に必要な入力ファイルの作成と計算の実行
・ 複数の入力ファイルを一括で実行できるバッチ計算用ツール
・ 反応解析に必要な遷移状態を反応物、生成物より計算する自動ツール
・ IRC計算の最終構造、エネルギープロファイルのCSV出力
Avocuteには、分子モデルをグラフィカルに表示する機能はありません。別途、分子モデリングのためのエディターを用意する必要があります。 Avocuteは、分子エディターにAvogadroを使用することを推奨しています。
Avogadroは、以下のページから無料で入手できます。
Avogadro
https://avogadro.cc/
【変更履歴】
・2020/11/06 Ver.2.0.0公開
・MOPAC6をMOPAC6.06へ変更
・mgfファイルの出力に対応(JSmolで分子軌道表示可能)
・メイン画面の[Calculation]にIRCメニュー追加
・メイン画面の[Options]の「VECTORS」を「GRAPH」に変更
・メニューバーのToolsに「IRC Checker」追加
・ARCファイルの読込みに対応
・JSmolのURL修正
・その他、軽微な不具合の修正
・2020/10/15 Ver.1.0.0公開
Avocuteの使い方
ダウンロードしたzipファイルを、適当なフォルダに解凍すればインストール完了です。解凍したファイルの中の「Avocute.exe」がソフトウェア本体になります。同梱の関連ファイルを削除したり、ソフトウェア本体をファイル外へ移動したりすると正常に動作しなくなるので注意してください。
Avocute.exeのショートカットを好きな場所に作成して利用することをおすすめします。
非常にシンプルなUIなので説明がなくても問題なく使用できるかと思います。
具体的な使用方法は以下の通りです。説明は、Ver.1.0.0の操作画面を用いて行っています。最新バージョンで追加された項目・機能については、その都度、補足します。
Avocute | メイン画面
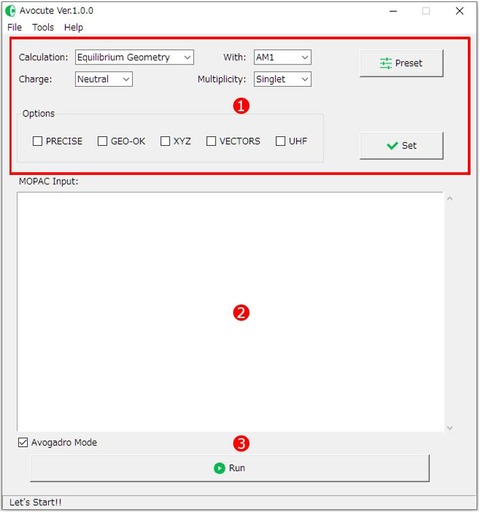
Avocute.exeを起動し、最初に表示されるのがメイン画面です。①が計算に必要な各種設定ボタン、②が入力ファイルのエディット画面(MOPAC Input:)、③[Run]が計算の実行ボタンです。
実際に「水分子の構造最適化計算」を例に実行方法を説明します。
構造最適化とは、簡単に言うと「分子の結合角や、結合長を含めた安定な構造を求める計算」のことです。
1)まず、Avogadroを起動し水分子を構築します。水分子の作成方法がわからない場合は以下のページを参照してください。
【関連記事】Avogadroを使ってみよう
http://pc-chem-basics.blog.jp/archives/396177.html
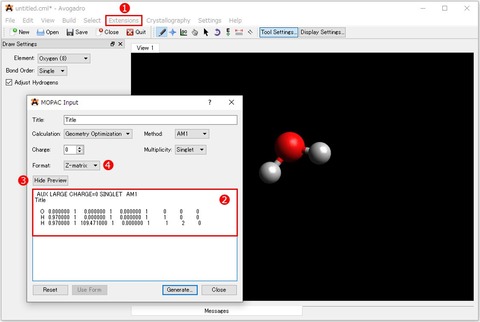
水分子が作成できたらAvogadroのツールバーから①「Extension>MOPAC...」を選択します。表示される「MOPAC Input」画面の②エディット画面のテキストを全て選択してコピーします。
エディット画面のテキストが表示されていない場合は、③[Show Preview]ボタンをクリックすることで表示/非表示(Hide Preview)を切替えることができます。 この画面で計算方法を指定しても当然問題ありません。作成例では、分子座標を④[Z-matrix]に指定し、その他の設定は変更しません。
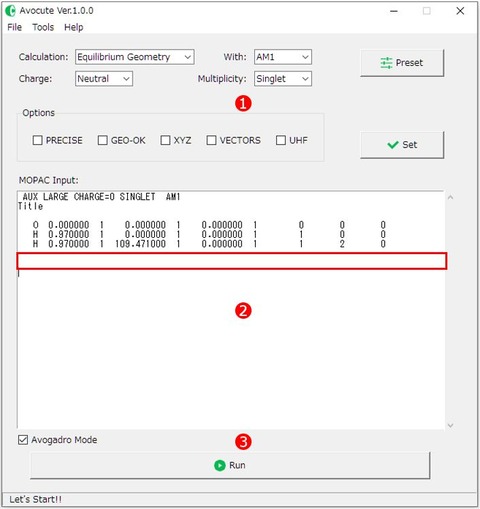
2)先程コピーしたテキストをAvocuteのメイン画面の②エディット画面(MOPAC Input:)に貼り付けます。 入力ファイルは、最初の行に使用するパラメータモデル(AM1, PM3など)と計算に必要なキーワードを指定します。 2行目と3行目はスペースです(個人的なメモを記入してもOKです)。4行目から、分子座標を入力します。そして、分子座標の最後に空白行という構成になっています。

貼り付けたテキストの最後に一行以上空白行があるか、必ず確認してください(赤枠部分)。これらはMOPACの入力ファイルの形式上の決まり事です。入力ファイルの形式が守られていない場合、計算エラーが出力されるので注意が必要です。
計算手法や分子座標について、さらに詳しく知りたい方は以下のページを参照してください。
【関連記事】
MOPACの入手とインストール方法
http://pc-chem-basics.blog.jp/archives/1496795.html
MOPACを実行してみよう
http://pc-chem-basics.blog.jp/archives/1509833.html
メイン画面のエディターは、直接書き込んだりコピペしたりする他にも、メニューバーの「File>Open Input File...(*.dat)」から既存のファイルを読み込むことが可能です。必要に応じて上手に使い分けてください。
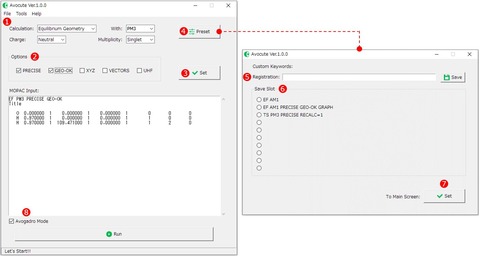
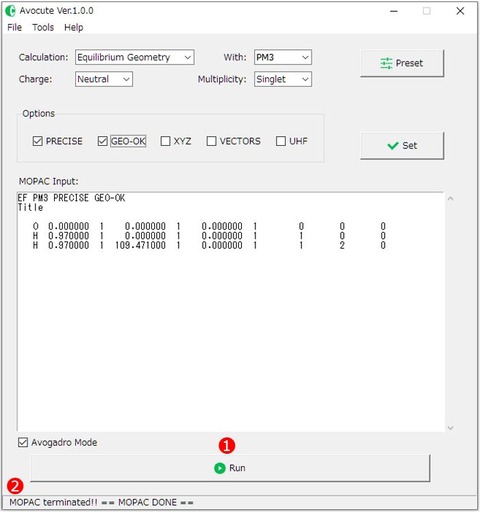
3)このまま実行ボタン[Run]をクリックして計算を実行することもできますが、ここでは設定ボタンで計算方法を変更してみましょう。使用例ではパラメータにPM3を用いて構造最適化計算を行います。つまり、各プルダウンメニュー①は図のように設定します。

CalculationはEquilibrium Geometry (構造最適化)、パラメータ(with)はPM3、電荷(Charge)、スピン多重度(Multiplicity)は中性分子なのでNutral、Singlet(一重項)をそれぞれ選びます。
これら4つは必ず適切に指定する必要があります。
オプション②は必ずしも必要ではありませんが、ここではPRECIESとGEO-OKにチェクを入れてみましょう。オプションに用意されているキーワードの意味は以下の通りです。
PRECISE:収束判定を100倍厳しくする (より厳密な構造最適化を希望する場合)
GEO-OK:原子が異常に接近した場合のチェックを無視する
XYZ: カルテシアン座標系で計算 (内部座標系でエラーが出る場合などに追加)
VECTOR:軌道係数、軌道エネルギーなどを出力ファイルに印字 (Ver.1.0.0のみ。Ver.2.0.0以降もキーワードは使用可能)
UHF:非制限ハートリー・フォック法 (開殻電子系に使用)
GRAPH:JSmolで分子軌道を表示するための書式付ファイル(*.mgf)を生成する (Ver.2.0.0より追加)
最後に③[Set]ボタンをクリックすれば、エディット画面の一行目に設定したキーワードが反映されます。
ちなみに、④[Preset]ボタンをクリックするとお気に入りのキーワードを登録して呼び出すことができます。⑤の入力欄に登録したいキーワードを入力して、保存したい⑥「Save Slot」を選んだ状態で右端の⑤[Save]ボタンをクリックすると登録完了です。⑦[Set]ボタンをクリックすればメイン画面にキーワードを反映することができます。
MOPACにはさらに多くのキーワードが存在します。MOPAC6に対応する全てのキーワードを確認するには、Avocuteのメニューバーの「Help>MOPAC6 Keywords」から参照してください。
メイン画面下、⑧[Avogadro Mode]のチェックが入っている場合は、MOPAC6に対応していないキーワード(AUX:他のプログラムで利用可能な追加情報ファイル*.auxの出力)を自動的に削除して計算します。これは、AvogadroのMOPAC入力ファイルのエディット画面で自動的にAUXが追加されていても、Avocuteでそのまま計算を実行できるようにするためのものです。キーワードAUXは最近のMOPACに実装されたものでMOPAC6には対応していません。
4)最後に、①[Run]ボタンをクリックして計算を実行してみましょう。はじめに入力ファル(*.dat)をどこに保存するかを選択すると、直ちに計算が実行されます。
保存先のディレクトリ名はスペースや特殊な記号などを含むとエラーが出るので、それらを含まない場所やファイル名で保存してください。Avocuteの実行ディレクトリも同様です。
計算が開始すると②ステータスバーの表示が「wait...」になります。計算が終了すると、「MOPAC terminated!!」と表示されます。 計算が正常に終了した場合は、「== MOPAC DONE ==」、エラーが発生している場合は「Error: Check the output!!」と続いて表示されます。
5)計算が正常に終了したら、入力ファイル(*.dat)を保存した場所に同じファイル名で「*.arc」、「*.out」が作成されているはずです。「*.out」が計算結果を記した出力ファイル、「*.arc」が「*.out」の要約を記したファイルになります。いずれも、テキストエディッタで開いて内容を確認することができます。計算エラーの場合は、「*.out」をテキストエディッタで開き出力ファイルに示されているエラーの原因を確認しましょう。
生成する「*.arc」、「*.out」は、すでに同じ名前のファイルが存在する場合、自動で上書きされます。他のツール(Batch Launcher、Auto TS Searcher)でもこの仕様は同様です。
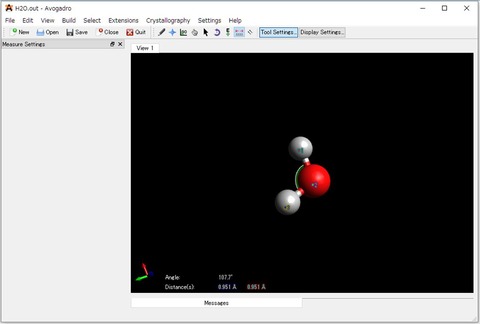
6)出力ファイル(*.out)は、Avogadroのメイン画面にドラックアンドドロップすることで構造最適化した構造を表示することができます。Avogadroを使えば、計算で得られた分子の結合長や結合角などをグラフィカルに確認することができます。
Avocute | Batch Launcher画面
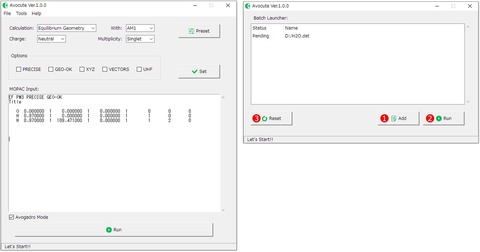
Avocuteのメニューバー「Tools」から「Batch Launcher」を選択すると、メイン画面で作成した入力ファイルを任意に登録して連続実行することが可能です。まず、メイン画面のエディターで入力ファイルを作成します。Batch Launcher画面の①[Add]ボタンをクリックすると入力ファル(*.dat)をどこに保存するか選択し、登録完了です。

あとは、同様に計算したいファイルを登録していき②[Run]ボタンをクリックすると計算が順次実行されます。計算が終了すると、Statusが「Pending」から「Done」に変わります。 登録したデータをクリアしたい場合は、③[Reset]ボタンをクリックしてください。
ちなみに、メニューバーの「File>Clear Temporary Files」を実行すると計算時に作成される不要な一時ファイルを削除することができます。通常の計算では自動的に不要な一時ファイルは削除されるので、このメニューを実行する必要はありません。 計算が途中でクラッシュした場合や、計算結果が以前の計算結果に影響を与えている可能性がある場合、またはAvocuteフォルダ内に不要なファイルが蓄積した場合などに実行してください。
Avocute | Auto TS Searcher画面
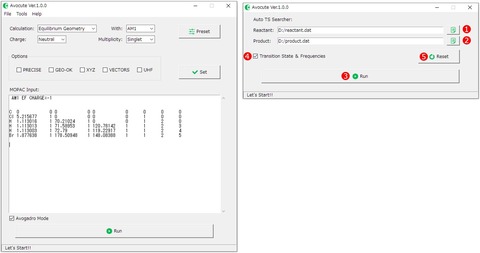
Avocuteのメニューバー「Tools」から「Auto TS Searcher」を選択すると、反応解析に必要な遷移状態を反応物、生成物より簡単に計算することが可能です。具体的にはMOPACの「SADDLE」という計算方法で遷移状態の候補構造を探索します。1)まず、メイン画面で反応物の構造最適化の入力ファイルを作成します。Auto TS Searcher画面の①Reactant(反応物) 登録ボタンをクリックし入力ファル(*.dat)をどこに保存するか選択し、登録完了です。同様にメイン画面のエディターで生成物の入力ファイルを作成したら②Product(生成物)登録ボタンをクリックし登録します。
2)この使用例では、Avocuteに同梱されている「SampleFile」フォルダの中から「product.dat」と「reactant.dat」を用いて計算を実行します。
サンプルファイルは塩化メチルと臭化物イオンの求核置換反応における反応物と生成物です。 登録が完了したら③[Run]ボタンをクリックして計算を実行しましょう。
④[Transition Stae & Frequencies]にチェックがある状態(デフォルト)では、SADDL計算後に遷移状態の最適化、および振動解析を実行します。チェックを外すとSADDLE計算で終了します。必要に応じて使い分けてください。
なお、Reactant(反応物)とProduct(生成物)の構造最適化後に行われる、SADDLE計算、遷移状態の最適化(TS)、振動解析(FORCE)で用いられるオプションのキーワードは、Reactant(反応物)の入力ファイル中に含まれるキーワードをそのまま継承します。
もし、登録したファイルを解除したい場合は、⑤[Reset]ボタンをクリックしてください。
3)計算が開始するとステータスバーの表示が「wait...」になります。計算が終了すると、「MOPAC terminated!!」と表示されます。 計算が正常に終了した場合は、「== MOPAC DONE ==」と続いて表示されます。
4)計算が正常に終了したら、反応物の入力ファイル(*.dat)を保存した場所に同じファイル名で「*.arc」、「*.out」が作成されているはずです(振動解析(キーワード:FORCE)の計算ではarcファイルは作成されません)。
遷移状態の候補化合物(SADDLE)は「反応物のファイル名_saddle」、その最適化計算(TS)は「反応物のファイル名_ts」、その振動解析は「反応物のファイル名_frq」というファイル名で保存されます。
5)出力ファイル(reactant_ts.out)を、Avogadroの画面にドラックアンドドロップすると計算で求めた構造をグラフィカルに表示することができます。

6)振動解析の結果(*_frq.out)はAvogadroで表示できません。グラフィカルに結果を確認したい場合は、Avocuteのメニューバー「Tools」から「JSmol」を選択するとWebブラウザでJSmolにアクセスできます。
ファイル選択メニュー①が表示されるので「reactant_frq.out」を選択して[load]ボタンをクリックすると目的の分子構造が表示されるはずです。
次に右クリックメニューの②「モデル」から目的の振動を選択します。ここでは、遷移状態の確認なので唯一存在する虚数振動(マイナスの振動数)を選択します。 最後に③「振動」をオンにすれば振動モードに対応したアニメーションが表示されます。
最後に、SADDLE計算をする際に注意しておきたいのは、内部座標系を用いる場合は反応物と生成物の各結合情報が一致していなければならないことです。例えば、反応前後で各原子に割り当てられる番号が異なっていたり、内部座標で定義されている参照原子との関係が異なったりしていると、エラーが出力されるので十分注意しておきましょう。
Avocute | 分子軌道の表示 (mgfファイル)
キーワードに「GRAPH」を用いた場合、保存したファイルと同じ名前の「*.mgf」ファイルが作成されます(ver.2.0.0以降)。このファイルは、JSmolで読み込むことで分子軌道の表示が可能です。Avocuteのメニューバー「Tools」から「JSmol」を選択するとWebブラウザでJSmolにアクセスできます。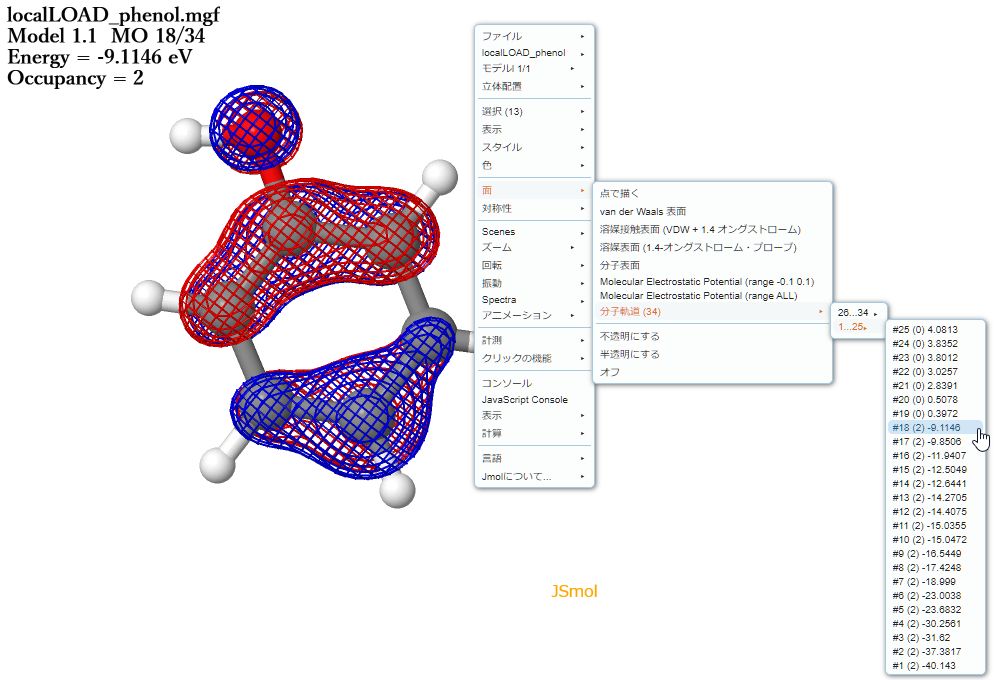
上図は、Avocuteに同梱されている「SampleFile」フォルダの中から「phenol.mgf」のHOMOを表示させた様子です。JSmolの右クリックメニューの「面>分子軌道」より目的の軌道を選択するだけです。
Avocute | IRC Checker
メニューバーの「Tools >IRC Checker」は、IRC計算の出力ファイルから、最終的な分子座標とエネルギープロファイルをCSVファイルへ書き出すことができます(ver.2.0.0以降)。IRC計算とは、得られた遷移状態が反応物と生成物を結ぶかどうかを確認するために用いられます。先述の「Auto TS Searcher」で得られた、遷移状態を元にIRC計算を行いながら一連の操作方法を説明します。
1) ここでは、Avocuteに同梱されている「SampleFile」フォルダの中から、「reactant_ts.arc」を用いて説明します。「reactant_ts.arc」には、計算で得られた、塩化メチルと臭化物イオンの求核置換反応における遷移状態の分子座標が含まれています。

メニューバーの「Fire >Open... >Arc Fire(*.arc)」からファイルを読み込みましょう。キーワードは、上図のように「IRC=1 LARGE=50 AM1 CHARGE=-1」と設定します。メイン画面の[Calculation:]のプルダウンメニューから「IRC」を選択した場合、「IRC=1 LARGE=50」がデフォルトです。
キーワード「IRC=1」は、虚の振動数をもつ基準振動の方向に構造変化させることを、「IRC=-1」は逆方向へ変化させることを意味します。遷移状態から、反応物と生成物への両方の確認が必要なので、「IRC=1」の計算が終了したら、「IRC=-1」に変更して計算を実行しましょう。ちなみに、「LARGE=50」は、50回ごとにIRC上の構造を出力する事を指定しています。「CHARGE=-1」は、モデル全体の電荷を指定しています。
2) 入力ファイルの作成が終了したら、メイン画面の[Run]ボタンをクリックして計算を行いましょう。計算が終了すると、保存したファイル名の「*.out」が出力されるはずです。
3) メニューバーの「Tools >IRC Checker」をクリックし、先程の計算で出力された「*.out」ファイルを読み込みましょう。IRC計算以外のOutputファイルや、計算が正常に終了していないファイルを読み込むとエラーが出力されます。

すべての処理が正常に終了すると、下図のように「Successfully terminated!!」というメッセージボックスが表示されます。また、読み込んだ「*.out」ファイルと同じ場所に、「*_ircf.out」「*_ircf_plot.csv」の2つのファイルが出力されます。
通常、IRC計算で得られる「*.out」ファイルをAvogadroで開くと、初期構造しか表示されません。一方、IRC Checkerで出力される「*_ircf.out」は、IRC計算で得られる最終構造を表示することができます。
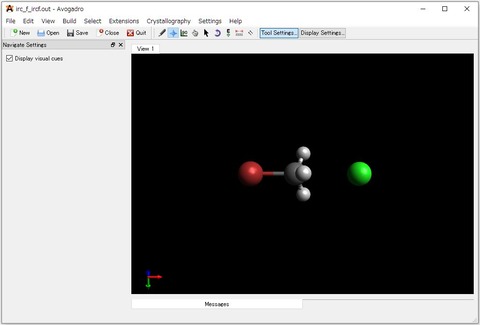
上図は、出力された「*_ircf.out」をAvogadroで表示させた様子です。IRC計算により、目的の生成物である臭化メチルと塩化物イオンが得られたことが確認できます。また、「*_ircf_plot.csv」は、エクセルなどで開き、A,B列を散布図でプロットすると、エネルギープロファイル図を作成することができます。
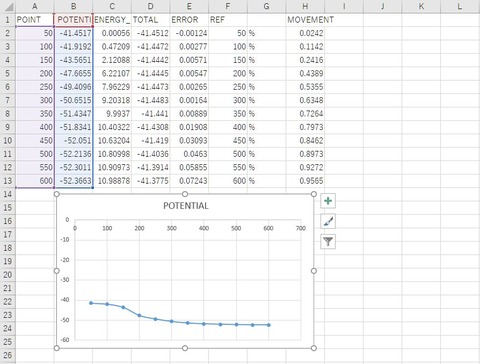

上図は、出力された「*_ircf.out」をAvogadroで表示させた様子です。IRC計算により、目的の生成物である臭化メチルと塩化物イオンが得られたことが確認できます。また、「*_ircf_plot.csv」は、エクセルなどで開き、A,B列を散布図でプロットすると、エネルギープロファイル図を作成することができます。

おわりに
Avocuteの原型は、数年前にMOPACのEF~SADDLE~TS~FORCE計算の一連の面倒な入力ファイルの作成から計算の実行をPythonで自動化するために作ったスクリプトです。最初は、「Pythonの標準(内部)ライブラリのみ」という条件で、このスクリプトを元に1週間でどの程度のソフトが作れるか「一週間チャレンジ」という縛りで製作を開始しました。今後は、少しずつ機能を追加していく予定です。
余談ですが、ブログの連載記事「Pythonではじめるアプリ開発」を一通り学べばAvocuteのようなソフトウエアを作成できます。また、書籍「Pythonではじめるデスクトップアプリ開発入門」では、さらに詳しいデスクトップアプリの開発方法を解説しています。興味がある方は、お手に取っていただければ幸いです!!
リンク

コメント
コメント一覧 (3)
このようなGUIがあるとチャレンジしてみようと思えますよね、素晴らしいと思います。
MDでは丁寧に解説しているブログや扱いが簡単なGUIに乏しい一方、
量子化学計算ではここのブログも含め、初心者向けの内容が豊富で助かっています。
興味があるのがPythonでのGUI作成なのですが、tkinter等を用いられたのでしょうか?
簡単なGUI作成は私もたまにやっているのですが、ここまでPythonで作成できることに驚き、
ちょっとスクリプトを見てみたいと思った次第です。
差し支えなければ、どんなスクリプトかを伺ってもよろしいでしょうか?
コメントありがとうございます。
GUIはお察しの通り、Tkinterで作っております。
外部モジュールなどは今のところ一切使っていません。
近日中にGitHub にソースコードを公開する予定ですので、もう少々お待ち下さい
また、他のユーザーさんからもTwitterでアプリケーションの作り方を知りたいとDM頂いていたので、初心者の方のために簡単なGUIの作成に関する記事を書く予定です。
これからも、よろしくお願いいたします。
tkinterで機能が充実したGUIを作成できるのですね。
今までtkinterは扱いが簡単な一方、機能は乏しいイメージがあったので、
これを機に勉強させて頂ければと思います。
GitHubでのソースコード公開、楽しみにしております。
よろしくお願いいたします。