
これまで、いくつかの量子化学計算パッケージで利用できるGUI(グラフィカル・ユーザ・インタフェース)ソフトを紹介してきました。お気に入りのソフトは見つかりましたか?
今回は、まだ紹介しきれていなかった無料で利用できる定番のGUIソフトを3つ紹介したいと思います。「特徴的な機能」に焦点をあてて紹介しますので、既に他のソフトを導入している方も、興味が湧いたら是非試してみてください。
ダウンロードはこちら < https://sites.google.com/site/allouchear/Home/gabedit/download >

結合角や結合長の表示はもちろん、分子軌道や静電ポテンシャルマップなどの分子表面の描画やIR, UV, NMRの表示など一通りの機能は備わっています。機能が豊富ですが、UIが少し古臭くて初心者には扱いづらいかもしれません。

これまで紹介してきたGUIソフトにない特徴的な機能としては、単位換算機能(ユニットコンバーター)と分子表面の描画に関する細かい設定が可能な所です。また、以前紹介したOpen Babelも内蔵しているので様々なフォーマットのファイル変換も可能です。

ツールバーから赤枠①「分子モデルの作成」、赤枠②「計算の出力ファイルの解析」、赤枠③「単位換算機能」にアクセスできます。Toolsからは、IR, UV, NMRの出力ファイルの読込みを行い描画させることができます。
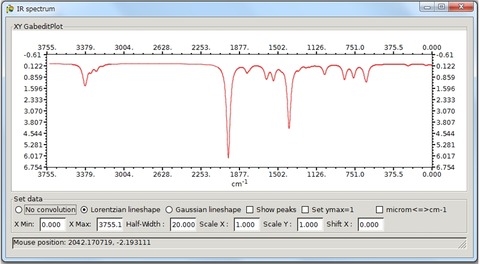
Tools >IR spectrumから以前計算したAcetophenonのIRスペクトルを表示させたのが以下になります。スケールファクターや細かい設定も可能です。
単位換算機能(ユニットコンバーター)は、a.u.からHartree、kcal/molからkJ/molなど計算化学でよく用いられる単位換算の機能が網羅されています。

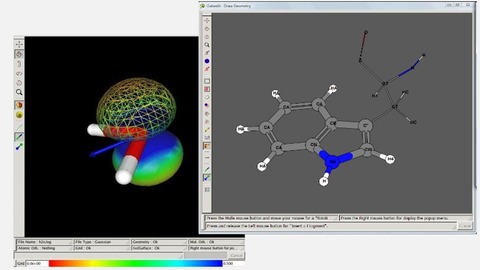
下図のように、分子表面の一方だけをメッシュ表示にしたり(左図)、特許の明細書でよく見るようなモデル表示(右図)も可能です。詳細設定は、分子モデルを描画させた画面の右クリックメニューから行えます。Mac版はコンパイルが必要なので、面倒な場合はWindows版をダウンロードしEasyWine上で動かすことをおすすめします。
ダウンロードはこちら < http://zzzfelis.sakura.ne.jp >

特徴的な機能としては、GAMESS(US)のフラグメント分子軌道(FMO) 法のインプットビルダーとして利用できるところでしょう。FMOは、系全体を小さなフラグメントに分割し、全電子計算を行います。小さな計算を複数回実行するだけなので、巨大な系を扱う際に計算コストを大きく減らすことが可能です。また、タンパク質のような巨大で複雑な分子の局所構造をみるためのLocal Structure Viewerや、フラグメント間相互作用を可視化する機能も備えています。
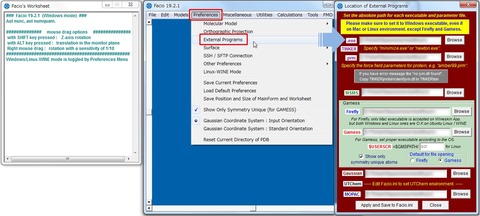
FacioでGAMESSなどのプログラムパッケージを使用する場合は、ツールバーのPreferences >External Programsからディレクトリを指定する必要があります。また、プログラムを実行する場合は、ツールバーの"Calculations"から目的のプログラム名を選択します。
計算した出力ファイルを読み込む場合は、File >Load New (プログラム名)から対応するOutputファイルかPUNCHファイル(DATファイル)を選択します。計算結果の可視化は、ツールバーのTools >Viewersから行います。
例えば、GAMESSで計算したIRスペクトルを表示させたい場合、「Punch(*.pun/*.dat) for Nomal Mode Vibration」からPUNCHファイル(DATファイル)とOutputファイルの両方を読み込む必要があります。

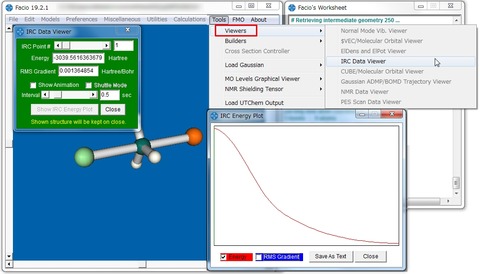
これまで紹介した、MaSKやMacMolPlt、そしてMoCalc2012の可視化に必要なのはOutputファイルだけでしたが、Facioの場合付属の出力ファイルも必要なので注意が必要です。GAMESSのIRC計算も下図のように視覚化することが可能です。
国産のソフトなので、日本語のマニュアルなどに簡単にアクセスできます。しかし、分子モデリングの方法や使い方が独特で、設定項目が細かいこともあり、初心者にとっては少々扱いづらいかもしれません。Facioは、Windows版とMac版がありますので興味のある方は試してみてください。
ダウンロードはこちら < http://www.chemcraftprog.com/lite.html >

フリー版は大きく機能が制限されているため、正直これといって特徴的な機能はありません。あえて挙げるなら、分子モデルの外観が独特で、カラフルといったところでしょうか。論文などでも良くChemcraftで作成した分子モデルを見かけます。Chemcraft Liteは無料版なので、起動するたびに毎回異なる6文字の英数字を入力する必要があります。
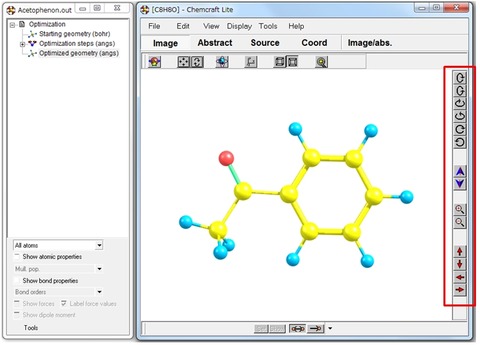
分子モデルの自由回転は、画面を右クリックしながらマウスを動かします。右のツールバーを利用して動かすことも可能です(赤枠)。結合長や結合角、電荷などの表示はできますが、分子軌道、エネルギー勾配および双極子モーメントの表示はできません(有料版は表示可)。Lite版の機能制限の詳細についてはHPを確認してみてください。
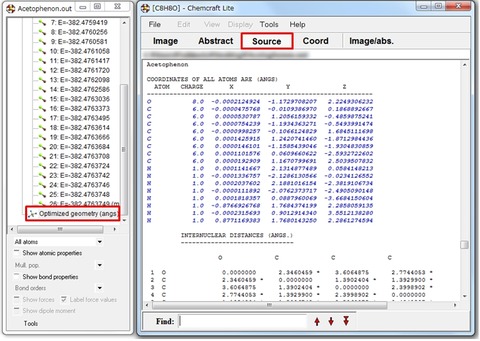
フリー版で利用できる機能のうち、唯一便利だなと思ったのは出力ファイルから目的の分子座標をin situで抽出できる機能です。上図のように、右側のサブウインドウから目的の計算結果を選択して、二段目のツールバーからSourceをクリックすると、選択した計算結果の分子座標が青色で表示されます。出力ファイルから目的の分子座標を抜き出す際には、便利な機能だと思います。
Chemcraft LiteはWindows版のみですので、MacユーザーはEasyWine上で動かしてみてください。もし、完全版を試用してみたい場合はトライアル期間が150日間あるので試してみると良いでしょう。
Chemcraft:http://www.chemcraftprog.com/download.html
※) 当ブログの連載が書籍になりました!!興味がある方は、お手に取っていただければ幸いです!!
(Amazon購入ページ)(書籍の紹介記事)

今回は、まだ紹介しきれていなかった無料で利用できる定番のGUIソフトを3つ紹介したいと思います。「特徴的な機能」に焦点をあてて紹介しますので、既に他のソフトを導入している方も、興味が湧いたら是非試してみてください。
Gabedit
Gabeditは、GAMESS (US)、Firefly(PC GAMESS)、Gaussian、MOLCAS、MOLPRO、MPQC、OpenMopac、ORCA、Q-chemなどの計算化学パッケージに対応する無料のGUIソフトです。ダウンロードはこちら < https://sites.google.com/site/allouchear/Home/gabedit/download >
結合角や結合長の表示はもちろん、分子軌道や静電ポテンシャルマップなどの分子表面の描画やIR, UV, NMRの表示など一通りの機能は備わっています。機能が豊富ですが、UIが少し古臭くて初心者には扱いづらいかもしれません。
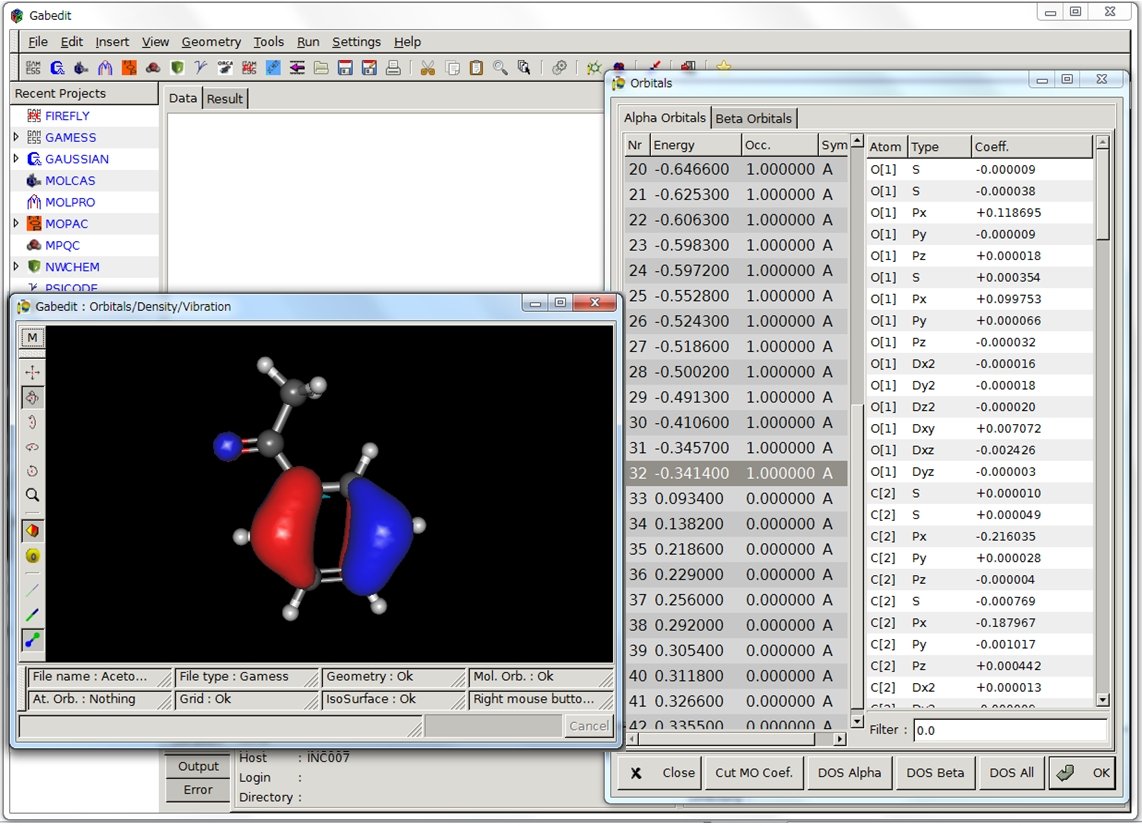
これまで紹介してきたGUIソフトにない特徴的な機能としては、単位換算機能(ユニットコンバーター)と分子表面の描画に関する細かい設定が可能な所です。また、以前紹介したOpen Babelも内蔵しているので様々なフォーマットのファイル変換も可能です。
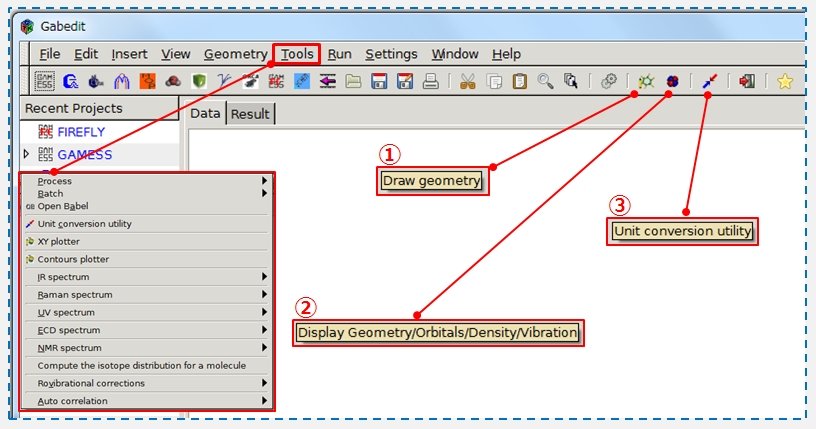
ツールバーから赤枠①「分子モデルの作成」、赤枠②「計算の出力ファイルの解析」、赤枠③「単位換算機能」にアクセスできます。Toolsからは、IR, UV, NMRの出力ファイルの読込みを行い描画させることができます。
Tools >IR spectrumから以前計算したAcetophenonのIRスペクトルを表示させたのが以下になります。スケールファクターや細かい設定も可能です。

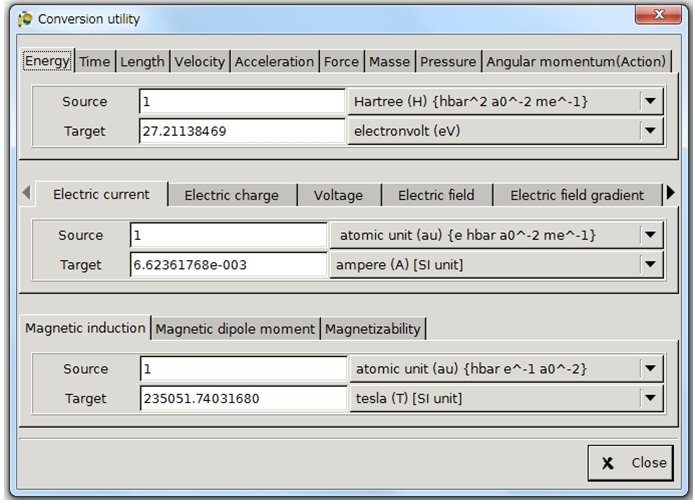
単位換算機能(ユニットコンバーター)は、a.u.からHartree、kcal/molからkJ/molなど計算化学でよく用いられる単位換算の機能が網羅されています。
下図のように、分子表面の一方だけをメッシュ表示にしたり(左図)、特許の明細書でよく見るようなモデル表示(右図)も可能です。詳細設定は、分子モデルを描画させた画面の右クリックメニューから行えます。Mac版はコンパイルが必要なので、面倒な場合はWindows版をダウンロードしEasyWine上で動かすことをおすすめします。

Facio
Facioは、GAMESS (US)、Firefly(PC GAMESS)、Gaussian、MOPAC、TINKER, MSMなどの計算化学パッケージに対応する無料のGUIソフトです。分子モデリングと計算の実行や可視化などの必要な機能は一通り備わっています。ダウンロードはこちら < http://zzzfelis.sakura.ne.jp >
特徴的な機能としては、GAMESS(US)のフラグメント分子軌道(FMO) 法のインプットビルダーとして利用できるところでしょう。FMOは、系全体を小さなフラグメントに分割し、全電子計算を行います。小さな計算を複数回実行するだけなので、巨大な系を扱う際に計算コストを大きく減らすことが可能です。また、タンパク質のような巨大で複雑な分子の局所構造をみるためのLocal Structure Viewerや、フラグメント間相互作用を可視化する機能も備えています。

FacioでGAMESSなどのプログラムパッケージを使用する場合は、ツールバーのPreferences >External Programsからディレクトリを指定する必要があります。また、プログラムを実行する場合は、ツールバーの"Calculations"から目的のプログラム名を選択します。
計算した出力ファイルを読み込む場合は、File >Load New (プログラム名)から対応するOutputファイルかPUNCHファイル(DATファイル)を選択します。計算結果の可視化は、ツールバーのTools >Viewersから行います。
例えば、GAMESSで計算したIRスペクトルを表示させたい場合、「Punch(*.pun/*.dat) for Nomal Mode Vibration」からPUNCHファイル(DATファイル)とOutputファイルの両方を読み込む必要があります。

これまで紹介した、MaSKやMacMolPlt、そしてMoCalc2012の可視化に必要なのはOutputファイルだけでしたが、Facioの場合付属の出力ファイルも必要なので注意が必要です。GAMESSのIRC計算も下図のように視覚化することが可能です。
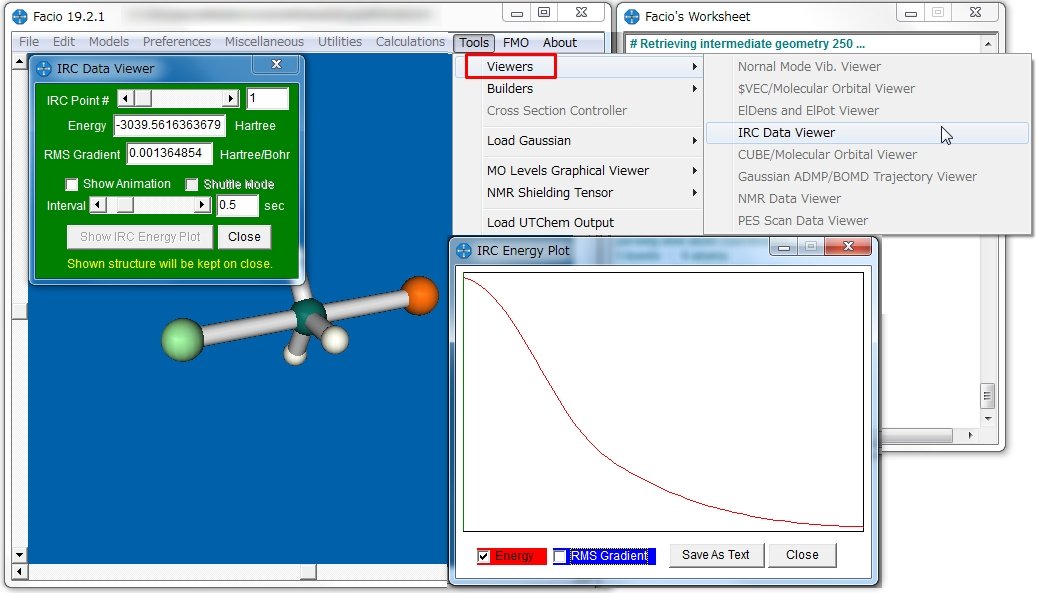
国産のソフトなので、日本語のマニュアルなどに簡単にアクセスできます。しかし、分子モデリングの方法や使い方が独特で、設定項目が細かいこともあり、初心者にとっては少々扱いづらいかもしれません。Facioは、Windows版とMac版がありますので興味のある方は試してみてください。
Chemcraft Lite
Chemcraft Liteは、GAMESS (US)、Firefly(PC GAMESS)、Gaussianなどの計算化学パッケージに対応するGUIソフトです。Chemcraftは有償ソフトですが、機能制限版のLiteはフリーウエアとして利用できます。ダウンロードはこちら < http://www.chemcraftprog.com/lite.html >

フリー版は大きく機能が制限されているため、正直これといって特徴的な機能はありません。あえて挙げるなら、分子モデルの外観が独特で、カラフルといったところでしょうか。論文などでも良くChemcraftで作成した分子モデルを見かけます。Chemcraft Liteは無料版なので、起動するたびに毎回異なる6文字の英数字を入力する必要があります。
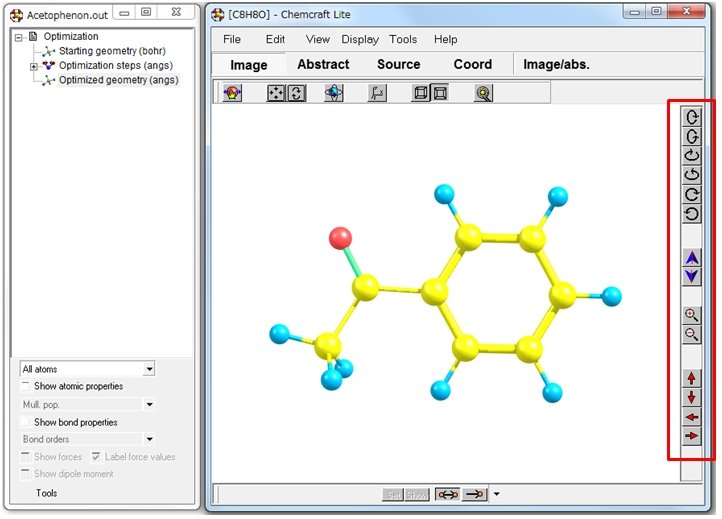
分子モデルの自由回転は、画面を右クリックしながらマウスを動かします。右のツールバーを利用して動かすことも可能です(赤枠)。結合長や結合角、電荷などの表示はできますが、分子軌道、エネルギー勾配および双極子モーメントの表示はできません(有料版は表示可)。Lite版の機能制限の詳細についてはHPを確認してみてください。

フリー版で利用できる機能のうち、唯一便利だなと思ったのは出力ファイルから目的の分子座標をin situで抽出できる機能です。上図のように、右側のサブウインドウから目的の計算結果を選択して、二段目のツールバーからSourceをクリックすると、選択した計算結果の分子座標が青色で表示されます。出力ファイルから目的の分子座標を抜き出す際には、便利な機能だと思います。
Chemcraft LiteはWindows版のみですので、MacユーザーはEasyWine上で動かしてみてください。もし、完全版を試用してみたい場合はトライアル期間が150日間あるので試してみると良いでしょう。
Chemcraft:http://www.chemcraftprog.com/download.html
おわりに
今回は、量子化学計算ソフトのGUIである「Chemcraft」, 「Facio」, 「Gabedit」の3つのソフトについて紹介しました。既に、「MoCalc2012」, 「MaSK」, 「MacMolPlt」, 「Avogadro」のいずれかを導入しているのであれば、必要性を感じないかもしれません。GUIに関しては、好みの問題が大きいので色々と試して自身で扱いやすいソフトを見つけてみてください。※) 当ブログの連載が書籍になりました!!興味がある方は、お手に取っていただければ幸いです!!
(Amazon購入ページ)(書籍の紹介記事)

コメント